
Mixed-modal Ligand in the Capture of Rat and Mouse Antibodies
Scott Conrad McCarthy, Logan Garrett, Irene Barber and Grant Shimamoto
Presented at American Chemical Society BIOT, New Orleans LA, 2014
Abstract
Rodent antibodies play a critical role during preclinical stages in development of biological therapeutics as surrogate and benchmark reagents. Often these benchmark biologics are scaled to meet large scale early preclinical in vivo trials. Although both Protein A and Protein G affinity methods are commonly employed to purify recombinant proteins containing rodent IgG domain, these processes have limitations as in low capacity, the inability to bind rat IgG2a subclass on Protein A, the increased acidity required to elute the biological proteins from Protein G, and the very high cost of Protein G resins. We have explored the use of mixed-mode resins as an alternative capture step to Protein G affinity chromatography for a variety of hybridoma rat and mouse antibodies. This study presents data using two mixed-modal two chromatography ligands, mercapto-ethyl-pyridine (4-MEP) and N-benzoyl-homocysteine, as having superior performance in a high throughput chromatography mode. Potential applications of these two ligands in improving the overall robustness of rodent antibody purification are discussed.
Keywords Rat monoclonal antibody; Mouse monoclonal antibody; Purification; Immunoaffinity chromatography; mixed-modal chromatography
1. Introduction
The production of rodent antibodies and rodent Fc fusion proteins are critical for the development of biological therapeutics. Production and purification of rat and murine antibodies are widely used in the validation of biological targets and for biophysical assays for patient samples. The first step in the investigation of a target is to develop models in rodent relevant to the biological effect in humans. The target protein and antibodies to the target protein are generated in multiple species to investigate the biological pathway. Diversity of antibody clones are evaluated for multiple epitopes and then tested for cross-reactivity to human biological proteins. The process of hybridoma production and screening has been significantly streamlined; however, it is often a challenge for the downstream separations group to handle the large numbers of these clones and to purify them to high quality.
Primarily at issue is that rodent antibodies bind poorly to Protein A [1,2], although somewhat better on recombinant Protein A than on natural Protein A. Protein A can be utilized to purify most rat and mouse species isoforms. The common capacity of recombinant Protein A for mouse and rat antibody is greater than 5 mg/mL with a few exceptions. Binding in a condition comprised of 0.75 M Glycine, 1.5 M NaCl, pH 8.9 condition can enhance the binding capacity for difficult clones as described by Shuler [3] and Bazin [4]. Most rodent antibody isotypes will bind strongly to Protein G; however, the capacities observed are similar to the capacity on Protein A with enhanced binding high salt pH 8.9. One exception is rat IgG2a which will not bind to Protein A. Protein G binding of our rodent antibody protein is approximately 5 mg/mL resin (Table 1). The benefit of Protein G is that the product is clarified in one purification step. The drawback includes the high cost of Protein G Sepharose, -especially in scaling the purification to 10 to 100 grams of protein which may be required for some studies. Also, the yield is low without using strong acidity which may denature the protein. In addition, we have found our columns may only be reused a limited number of times due to weak tolerance to the more stringent cleaning solutions [5].
The use of hydrophobic charge-induction chromatography, or mixed-modal chromatography (MMC), has been widely approached in capture of antibody protein over the last few years. The compelling reason to use mixed modal chromatography for the capture of antibodies is because traditional immunoaffinity chromatography is expensive and susceptible to degradation. Mixed-modal chromatography has properties of higher throughput and more robust cleaning between production runs than what can currently be used for Protein G. We report the binding profile of five model monoclonal antibodies onto Protein G and Protein A. We then compare this with the evaluation of multimodal ligand capture on mercapto-ethyl-pyridine (4-MEP) and Nbenzoyl-homocysteine (Capto MMC)[6,7]. 2.
2. Methods
Experimental 2.1. Materials
A selection of 5 model proteins of different rat and mouse isoforms were utilized in this study: rat IgG1, rat IgG2a, mouse IgG1, IgG2a and IgG2b. Spleen cells from immunized mice and rats- immunized against irrelevant protein- were fused to mouse myeloma cells. The hybridoma cells lines were maintained in BD Cell Mab Quantum Yield medium (BD Biosciences, San Jose, CA) with 10% fetal bovine serum and antibiotics at 37C, 10% CO2, in a humidified incubator. The cells were cultured and expanded in T175 flasks by splitting them at a ratio of 1:2. Cell density and viability were monitored using Vi-Cell automated cell counter (Beckman Coulter). During production the cells were spun at 1000 rpm and re-suspended in BD Cell Mab Quantum Yield medium with or without 10% super low IgG fetal bovine serum (Hyclone, Thermo Fisher Scientific, Rockford, IL) and antibiotics. Roller bottles were seeded at 5x10e5 cells per ml, filled with 10% CO2 gas, sealed and placed on roller racks in a 37C incubator. Every two days the bottles were again filled with 10% CO2 gas, glutamine and glucose were added during this time when needed. The roller bottles were maintained at 37C for 9 days before the culture medium was harvested [5]. Expression titers were determined using Forte Bio Octet® (Pall Corporation, Port Washington, NY) Protein G sensor.
2.2. Chromatography equipment
Chromatography experiments were carried out using an AKTA 100 Explorer FPLC system from GE Healthcare (GE Healthcare, Piscataway, NJ) measuring A280 nm for peak collection. Sodium chloride, sodium acetate, L-arginine, MES monohydrate, potassium phosphate, and TRIS base buffers were prepared from material obtained from J.T Baker (Center Valley, PA). Chromatography media was packed by hand in Tricorn™ column housing at 2 and 5 mL column bed volumes (GE Healthcare, Piscataway, NJ).
2.3. Immunoaffinity
chromatography
Protein A affinity chromatography was carried out using media obtained from two common commercially available sources: Mab Select™ (GE Healthcare, Piscataway, NJ) and POROS® 20 A (Applied Biosystems, Foster City, CA). The antibody was applied to the resin in 25 mM Tris, 150 mM NaCl, pH 7.4 and then washed in the same buffer. Typical elution was in 100 mM acetate pH 3.5. The eluates were immediately titrated to neutral pH using 1 M Tris pH 8.0. Protein G coupled media was obtained from two different common commercially available sources: Protein G Sepharose (GE Healthcare, Piscataway, NJ) and Protein G Plus UltraLink® (Thermo Fisher Scientific, Rockford, IL). The antibody was applied to the resin in 25 mM Tris, 150 mM NaCl, pH 7.4 and then washed with the same buffer. The typical elution condition was in 125 mM acetic acid, pH 2.7. The eluates were immediately titrated to neutral pH using 1 M Tris pH 8.0 [8,9].
2.4. Mixed-modal chromatography For strong anion exchange MMC Hypercell™ MEP was used (Pall Filtron, Port Washington, NY); for weak cation exchange MMC we obtained Capto™ MMC, Nbenzoyl-homocysteine coupled sorbent, (GE healthcare, Piscataway, NJ). Binding and elution conditions were initially screened using 96 well filter plates from Whatman. In 250 uL solution 50 uL of bead with 250 ug pure antibody was incubated for two hours modifying the pH and salt condition on each sample and sorbent type. Buffer stocks were formulated at 500 mM solution in sodium acetate at pH 3.5 to 5.0, MES monohydrate at pH 5.5 to 6.5, potassium phosphate at pH 7.0 to 7.5, and TRIS base at pH 8.0 to 8.9. Sodium chloride stock was formulated at 1 M solution. The resin was washed two times in the appropriate salt and buffer and the flow through retained by filtration by centrifugation and A280nm absorbance read on the a plate reader. The antibody was the eluted from the resin bead using 100 mM Acetic Acid pH 3.0 for 4-MEP or using 1 M NaCl, 50mM Tris, pH 8.0 for Capto MMC. For elution screening the antibody was bound in 25 mM K3PO4, pH 7.5 for 4-MEP and 25 mM MES pH 6.0 for Capto MMC. The bead was then washed in the appropriate binding buffer and eluted at various pH sodium chloride concentrations [10]. For column chromatography hybridoma supernatant was adjusted to 25 mM K3PO4, pH 7.5 and run on 4-MEP at a linear flow rate of 3 cm/min and eluted in 100 mM sodium acetate pH 4.0. For Capto MMC the supernatants were adjusted to 25 mM MES pH 6.0 and eluted using 600 mM NaCl in K3PO4, pH 7.6. Conditions for the optimization binding and elution were carried out in several buffers and salt conditions and at various pH levels [6].
2.5. Orthogonal chromatography
The second step purification was carried out on cation exchange or ceramic hydroxyapatite. CHT™ Ceramic Hydroxyapatite type 1 (HA) from Bio-Rad (Hercules, CA) was used binding in 10 mM potassium phosphate at pH 6.8, eluted in a linear gradient from 0 to 1 M sodium chloride and cleaned using 2 M sodium hydroxide. Flow rate was 1cm/min [10,11]. Cation Exchange (CEX) was carried out Fractogel EMD SO3 M (Merck Millipore, Darmstadt, Germany) using 5 mL columns. Binding was accomplished diluting the antibody in 20 mM acetate pH 5.0 and eluting step wise in 20 mM acetate 0.8 M sodium chloride pH 5.0 [6]. and Superdex200 size exclusion chromatography (SEC) (GE Healthcare, Piscataway, NJ). 2.6. Size exclusion chromatography Polishing and formulation of antibody product was achieved using Superdex200 size exclusion chromatography (SEC) (GE Healthcare, Piscataway, NJ) or Prepscale tangential flow filtration (Merck Millipore, Darmstadt, Germany) in Cellgro® PBS pH 7.2(Corning, Manassas, VA).
2.7. Protein titer and concentration determination
Titer of antibody in cell culture media and concentration of protein in the flow through were determined using Forte Bio Octet® (Forte Bio, Menlo Park, CA) against purified isotype standards with Protein G sensors. Concentration of purified antibody were read by NanoDrop ND1000 spectrophotometer (Thermo Scientific, Wilmington, DE) using an extinction coefficient of 1.5 absorbance cm-1 for 1 mg/mL at 280 nm in water.
2.8. Analytical size-exclusion chromatography
Comparison of aggregate profile was carried out using Analytical size-exclusion chromatography (SEC) on UPLC using the Acuity UPLC® BEH200 SEC 1.7 um 4.6 x 150 mm column (Waters Corp., Milford, MA). The injection volume was normalized to 10ug protein in 100 mM NaH3PO4 with 200 mM NaCl at a pH of 6.8 and volumetric flow rate of 0.3 mL/min. The absorbance was measured at 280 nm.
2.9. SDS-PAGE analysis The process intermediates and purified antibody sample was analyzed by Novex® 4-20% Tris-Glycine gel electrophoresis (Life Technologies, Grand Island, NY). The sample was diluted 1:1 in Tris sample buffer containing 30 mM iodoaceticmide for non-reducing condition or 25 mM DTT for reducing condition. The samples were heated for 10 min at 70°C and 5ug protein was added to each well. Protein bands were stained using Instant Blue (Accurate Chemical and Scientific Corporation, Westbury, NY).
2.10. Capillary Isoelectric Focusing
Capillary Isoelectric Focusing (i-CIEF) analyses were performed on an iCE280 system (Convergent Bioscience Ltd., Toronto, Ontario, Canada) equipped with a PrinCE Microinjector autosampler (Prince Technologies, the Netherlands). The separations were carried out on a short fused-silica column cartridge coated with fluorocarbon (50 mm Lx 100 µm inner diameter). The catholyte was 100 mM NaOH in 0.1% MC and the anolyte was 80 mM H3PO4 in 0.1% MC. Samples were introduced into the cartridge by autosampler held at 8oC and transferred to the cartridge by pressure and were focused for 1 min at 1500 V first followed by 6-10 min at 3000 V based on individual samples. Protein was detected at 280 nm and then was captured by a CCD camera. Data were analyzed by an iCE 280 Convergent Bioscience Ltd. (Toronto, Ontario, Canada) and Empower 2 Chromatography Data Software (Waters Corporation, Milford, MA, USA).
2.11. Differential Scanning Calorimetry
Differential scanning calorimetry (DSC) measurements were obtained using a VPCapillary DSC system (Microcal, Inc., Northampton, MA). Protein samples were diluted to 0.5 mg/mL while the corresponding buffer was used as a reference. The samples were scanned from 20 to 95 oC at a rate of 60 oC/h with an initial 20 min of equilibration at 20 oC. The data were analyzed using Origin 7.0 software (OriginLab1 Corporation, Northampton, MA). Thermograms were corrected by subtraction of bufferonly blank scans. The corrected thermograms were normalized for protein concentration. The melting temperatures, Tm, was determined using the DSC functions built into the Origin 7.0 software.
3. Results and Discussion
3.1. Protein G
In contrast to the capture and purification of human therapeutic antibody the hybridoma antibody titer in culture tends to be low. The host contaminant levels are high in ratio to the target mouse or rat antibody. The control which was used to compare product of alternate rodent antibody capture was the purification of our five model proteins on Protein G. Immunoaffinity chromatography offers high selectivity of the rodent monoclonal antibodies even at low concentration in the production media. The binding affinity of the rodent antibody protein was determined using binding onto a 1 mL Protein G column by the pH elution strength required to achieve better than 90% yield. It can be seen in Figure 1 that a much stronger acidic condition without salt was required to obtain yields of 90% [12]. The result set (Fig. 1) is typical of what is found using common elution strengths for Protein G. Elution using conditions below pH 3.0 can result in antibody aggregates forming from denaturation of the protein as is demonstrated on the rat IgG1 (Fig. 2). A stringent acetic acid condition of 125 mM or higher at pH 2.7, without added salt from titration, were required to achieve yields of 80% recovery of the five hybridoma test antibodies. As long as the proteins elute into a 1 M Tris pH 8.0 neutralizing solution aggregates are typically below 2% of the total product.
3.2. Mixed Modal Resin Screen
The benefit sought from mixed-modal chromatography capture was a robust process which would provide scalability with comparable product purities to traditional affinity purification without using the harsh acidic conditions required for protein G affinity chromatography. Three common commercially available mixed-modal ligands coupled sorbents were tested in the lab- two of which worked well for rat IgG1 and IgG2a antibodies: mercapto-ethyl-pyridine (or 4-MEP) and N-bezoyl-homocysteine (or Capto MMC) (Fig. 3). The primary interest in these ligands is for the purification of research rat IgG2a isoform antibodies which can only be purified on Protein G and cannot be captured using Protein A. From the plate based screening techniques, utilized to gain a better understanding of the salt tolerance and optimal pH for binding the various irrelevant rat Isotype control sample proteins, standard binding and elution conditions were determined for each MMC sorbent. The target range for binding onto 4-MEP appear to be non-dependent on conductivity and better in the range of pH 6.5 to 7.5. The binding of rat antibody on Capto MMC is better from pH 5.0 to 6.5 and better below 100 mM NaCl. Based on the trends seen from the binding plates, a binding condition of 25 mM K3PO4, 50 mM NaCl, pH 7.5 was chosen for 4-MEP and 25 mM MES, 50 mM NaCl, pH 6.0 was chosen for Capto MMC. The model rat proteins eluted from 4-MEP below pH 4.5 and below 100 mM sodium chloride. The proteins eluted from the weak ion exchanger at a pH above 7.0 and 400 mM sodium chloride (Fig. 4).
3.3. Mixed-modal Ligand Chromatography
Targeting a 20 mg/mL resin load capacity using purified rodent antibodies on column chromatography the results show promise toward the goal of developing a global method which would work for the majority of rodent antibody isotypes. The apparent dynamic binding capacity for 4-MEP was approximately 13 mg/mL resin and greater than 15 mg/mL resin for Capto MMC. The binding for the mouse IgG2b antibody tested was poor. Other mouse IgG2b Isotype proteins have in the past shown binding results comparable to the model mouse IgG1 and IgG2a results given in Table 2. The pI by cIEF for the mouse IgG1 and IgG2a are above 7 which typical of most hybridoma antibody clones. The pI for the mouse IgG2b used in this study was found to be 5.5 which would likely explain the poor binding observation on mixed modal ligand chromatography in our chosen pH binding condition. The binding of the hybridoma culture supernatants on 4-MEP or Capto MMC was targeted a capacity of 10 mg/mL resin based on the titer information. The observed capacity determined by Forte Bio compared well to the pure antibody binding experiments. It is important to note that the initial hybridoma supernatants tested contained 5% serum after harvest. Subsequent productions were carried out using serum free production. This was due to the fact that some BSA did bind to the column although most of the serum albumin from the serum productions flowed through on the two of the different MMC sorbents. Low levels of free light were also observed in the protein G affinity chromatography step. Higher levels of light chain domain were seen in the elution from the two MMC purifications. The addition of a pH 4.0 elution condition cleared a significant portion of the light chain and media components captured by 4-MEP [13] and a 100 mM NaCl wash was utilized on Capto MMC to clear free light chain. Results for single step capture for 4-MEP and Capto MMC are tabulated in Table 2. The initial capture step purity on MMC as assessed by analytical SEC is lower than traditional Immunoaffinity chromatography with the primary contaminant being the product related free light chain. Surrogate test material is routinely purified under multiple orthogonal column steps in order to achieve the high purity requirements which are meant to mimic therapeutic preclinical test material QC profiles. For smaller scale screens the chromatographic process of 4-MEP followed by CEX, and Gel-Filtration Chromatography (GFC) have been used to purify 20 - 100 mgs. The final step of gel filtration is commonly used to formulate the proteins at this scale. For the rat IgG2a on MMC, which was the focus of the experiments, it was found to compare well against the Protein G three step process by purity and yield (Table 3). For the larger purifications MMC followed by CEX and then tangential flow filtration for formulation have been used for gram amounts of antibody.
4. Conclusion
Mixed-modal Ligand Chromatography has been demonstrated to be an acceptable form of capture of rodent antibody protein. For smaller scale screening of clones the antibody can be typically polished using IEX followed by gel filtration for formulation. For the mid to larger scale productions, multimodal capture strategies can still be applied by polishing with hydroxyapatite and formulation using tangential flow filtration systems (TFF). Recombinant Protein A is currently used to purify large numbers of surrogate antibodies in the research setting. There have been a small number of murine monoclonal antibodies which have benefited significantly in binding to Protein A from the high salt and pH protocols. The high salt is not effective in promoting the binding of the rat IgG2a sub-isotype. Protein G is still commonly used to capture rat antibodies. The denaturation-induced aggregates from the Protein G elution step can be mitigated upon immediate neutralization using 1 M Tris, pH 8.0. 4-MEP provided approximately 13 mg/mL resin binding and met targeted yield and acceptable purity level of a modest expression hybridoma antibody. This was significantly better than the protein G response at half the residence time and provided a gentle alternative pH elution condition. Capto MMC provided higher capacity than Protein G and provides an alternate strategy of eluting in a modest salt condition rather than low pH buffer. Three step purification of the test proteins overall compared well to affinity purified antibodies as demonstrated by SDS-PAGE gel, differential scanning calorimetry (DSC) (Fig. 5) and analytical SEC analysis. In some cases with yield and purity better than our affinity purified controls.
Acknowledgements
We would like to thank Min Shen and Vladimir Razinkov for biochemical analysis. Mike Brown for chemical structure. Kim Shigenaka for hybridoma cell lines and instruction. Previous separations of rat IgG2a on 4-MEP have been prepared by Barbara Tipton.
References
1. P. Gagnon, Purification Tools for Monoclonal Antibodies, Validated Biosystems Inc., Tucson, Arizona, 1996.
2. E. Harlow, and D. Lane, Antibodies: A Laboratory Manual, Cold Springs Harbor, New York, 1988, pp. 617.
3. G. Shuler, and M. Reinacher, J. Chrom. A. 587 (1991) p. 61.
4. H. Bazin, J. Malache, J. Immunological Methods, 88 (1986) p. 19.
5. B. Owuor, E. Remarque, B. Faber, A. Thomas, J. Immunological Methods, 352 (2010) p. 192.
6. S. Hofer, A. Ronacher, J. Horak, H. Graalfs, W. Linder, J. Chrom. A. 1218 (2011) p. 8925.
7. R. Majors, A. Lees, M. Burkhardt, LCGC North America, 27 (2009) p. 14
8. S. Ohlson, R. Nilsson, U. Niss, B. Kjellberg, C. Frieburghaus, J. Immunological Methods, 114 (1988) p. 175.
9. S. Kabir, J. Med. Microbiol. 34 (1991) p. 167.
10. C. Morrison, P. Gagnon, S. Cramer, J. Chrom. A. 1217 (2010) p. 6484.
11. P. Gagnon, C. Cheung, E. J. Lepin, A.M. Wu, M. A. Sherman, A. A. Raubitschek, and P.J. Yazaki, Bioprocess Int. 8 (2010) p. 26.
12. G. Gaza-Bulseco, S. Faldu, K. Hurmans, C. Chumsae, H. Liu, J. Chrom. B. 870 (2008) p. 55. 13. H. Tong, D. Lin, X. Yuan, S. Yao, J. Chrom. A. 1244 (2012) p. 116.
Images
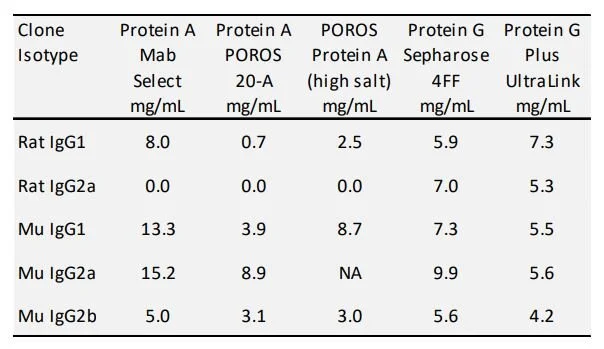
Table 1. Dynamic binding capacity model antibodies.
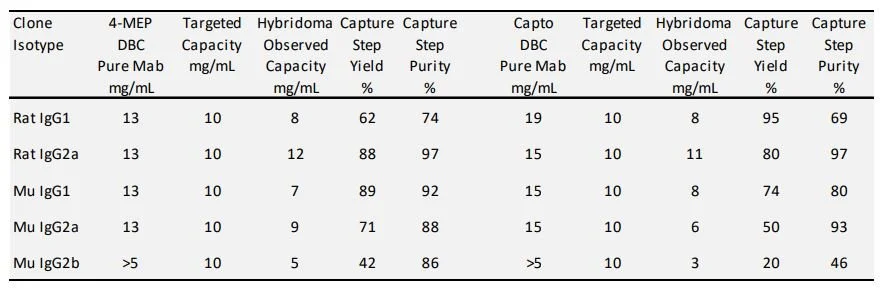
Table 2 Capture step hybridoma production media on 4-MEP and Capto MMC.
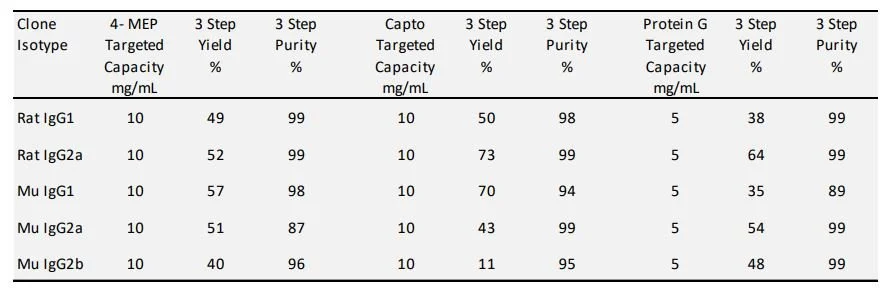
Table 3. Three step purification of hybridoma production media on 4-MEP, Capto MMC and Protein G coupled resin.

Fig. 1. Commonly used elution buffers for Protein G on three model rodent antibodies. Antibody was bound at 3 mg/mL resin at 5 min residence time then quickly neutralized upon elution.
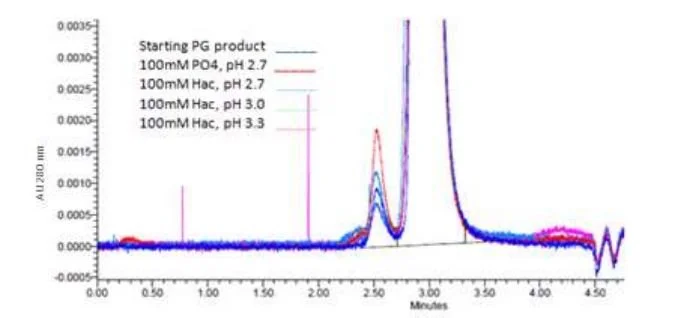
Fig. 2. Denaturation-induced aggregates from the Protein G elution step by UPLC Analytical SEC Assay. The amount of aggregation increases with low pH and increased acid strength.
Fig. 3. Structures of A) mercapto-ethyl-pyridine (4-MEP) a strong anion exchanger and B) Nbenzoyl-homocysteine (Capto MMC) a weak cation exchanger.
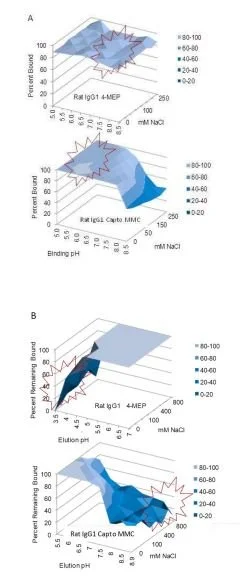
Fig. 4. A) Binding and elution profiles for rat IgG1 from 96 well filter plate study. Purified rodent antibody was bound to 4-MEP or Capto MMC beads at 250 ug Ab per 50uL resin. A) Binding conditions in 25 mM buffer from pH 5.0 to 8.5 in 0 - 250 mM NaCl. B) Elution conditions binding Mab onto 4-MEP in 25 mM K3PO4, pH 7.5 or 25 mM MES pH 6.0 onto Capto MMC. Buffer pH was varied from 3.5 to 7.0 on 4-MEP and pH 5.0 to 8.5 and 0 to 800 mM NaCl for Capto MMC.
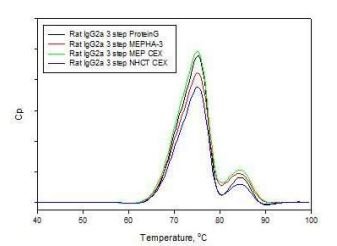
Fig. 5. DSC profiles of rat IgG2a antibody products purified from serum free hybridoma media captured on Protein G, 4-MEP, or Capto MMC, followed by CEX or HA and formulated into PBS on gel filtration chromatography.